为了查看降维聚类的可视化效果,我们先用相似样本降维聚类,然后使用具有差异的样本查看聚类效果。
同时使用 PCA 与 TSNE 来观察两种不同方法的聚类效果。
rm(list=ls())
library(pheatmap)
library(Rtsne)
library(ggfortify)
library(mvtnorm)
# 生成数据(随机)================================
# 设置基因数和细胞数
gene_num=100
cell_num=50
# 设置两个正态分布的随机矩阵(100*50) 作为两个样本矩阵
sample1=rnorm(gene_num*cell_num);
# 生成 100 基因,50 细胞的表达矩阵
dim(sample1)=c(gene_num,cell_num)
# 生成第二个样本
sample2=rnorm(gene_num*cell_num);dim(sample2)=c(gene_num,cell_num)
# 将两个样本合并为一个表达矩阵
sample_all=cbind(sample1,sample2)
# 添加列名
colnames(sample_all)=c(paste0('sample1_cell_',1:cell_num),
paste0('sample2_cell_',1:cell_num))
# 添加行名
rownames(sample_all)=paste('gene_',1:gene_num,sep = '')
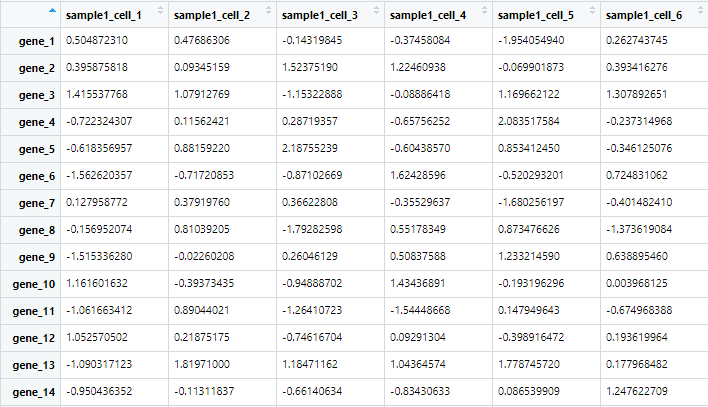
pheatmap(sample_all)
可以看到,由于数据随机分布,绘制热图发现两种数据并没有明显分组。
# 画pca===============================================
# 转置,让行为样本
sample_all=t(sample_all)
# 添加样本分组信息
df=cbind(as.data.frame(sample_all),group=c(rep('group1',20),rep('group2',20)))
# prcomp()主成分分析
pca_dat <- prcomp( df[,1:(ncol(df)-1)] )
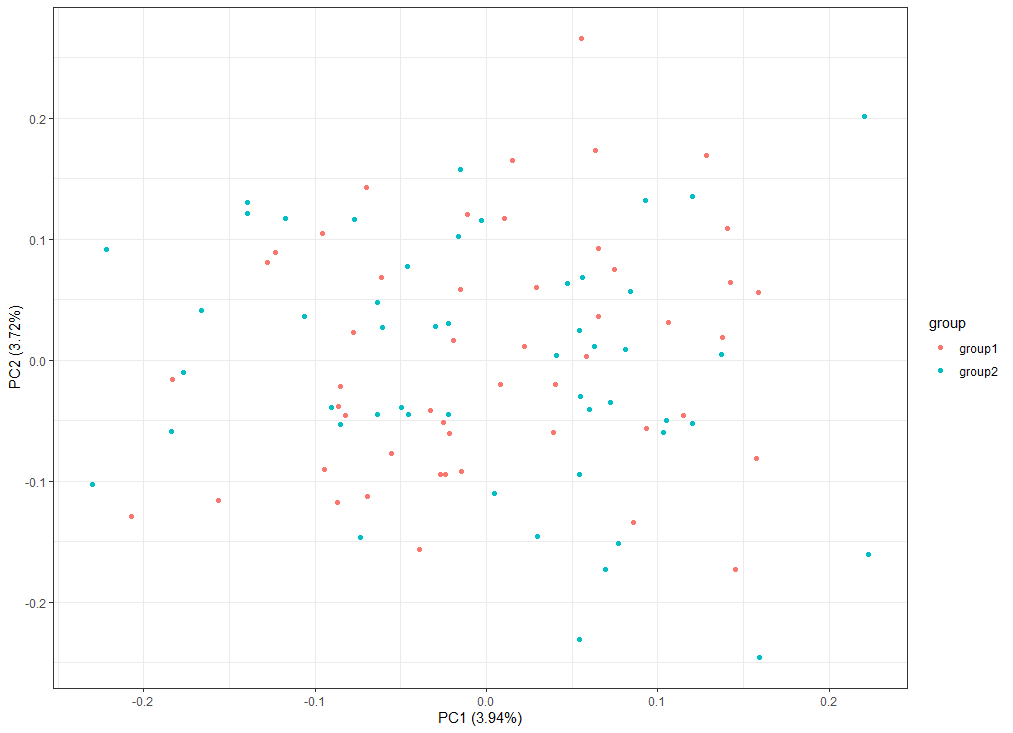
autoplot(pca_dat, data=df,colour = 'group')+theme_bw()
同样可以看到,两组数据的随机分布,导致组间数据并不能有效的分为两群,而是你中有我,我中有你的状态。
set.seed(123123)
sample_all=t(sample_all)
tsne_out <- Rtsne(sample_all,pca=FALSE,perplexity=10,theta=0.0)
# 获取tSNE的坐标值
str(tsne_out)
# 其中在Y中存储了画图坐标
tsnes=tsne_out$Y
colnames(tsnes) <- c("tSNE1", "tSNE2") #为坐标添加列名
# 在此基础上添加颜色分组信息,首先还是将tsnes这个矩阵变成数据框,然后增加一列group信息,最后映射在geom_point中
tsnes=as.data.frame(tsnes)
group=c(rep('group1',cell_num),rep('group2',cell_num))
tsnes$group=group
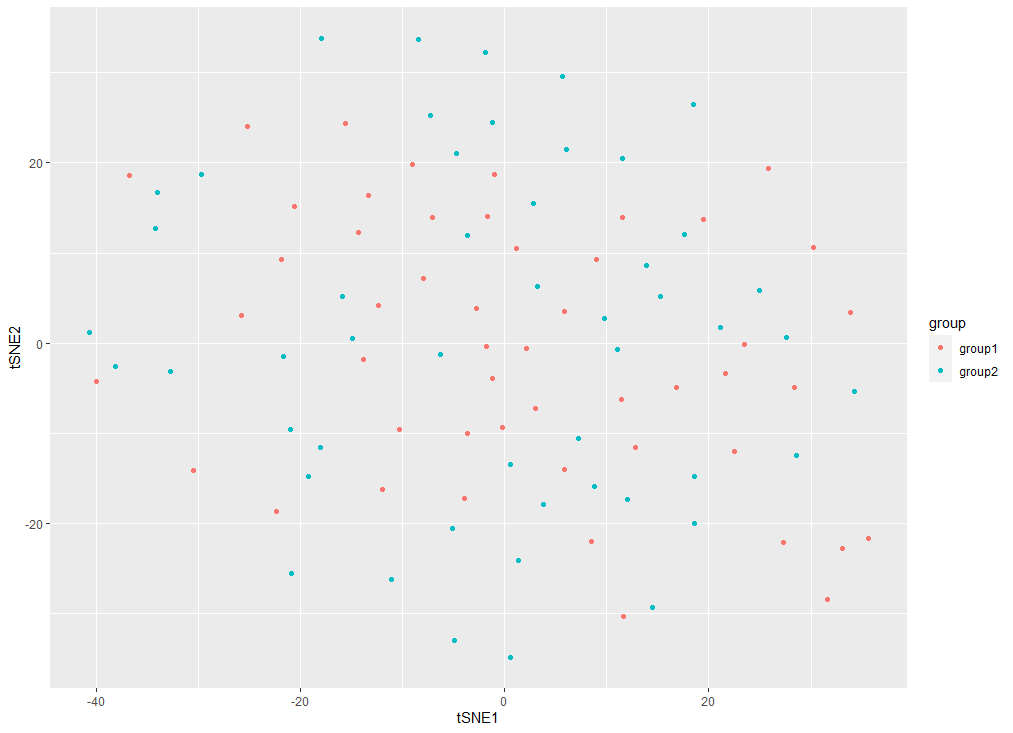
ggplot(tsnes, aes(x = tSNE1, y = tSNE2))+ geom_point(aes(col=group))
TSNE 与 PCA 是同样的展示效果,也就是说如果分析数据的变量之间没有明显的区别,利用不用的聚类手段或可视化方法,我们也不能将其分为不同群体。
在机器学习中,我们可以利用一些特征挑选手段实现相似数据间的分类,今后再讨论这种情况。
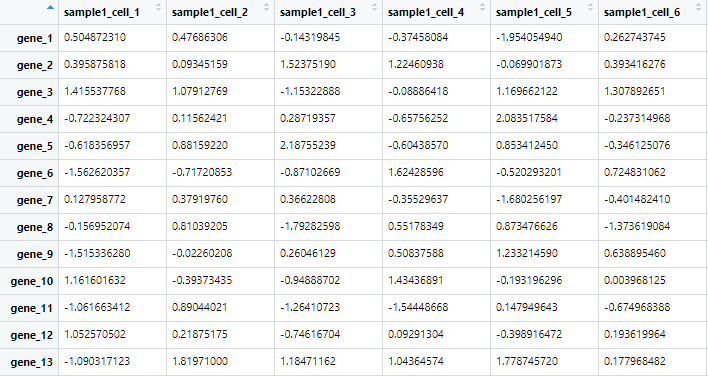
为了查看并比较数据间的差异,我们构建一个随机分布,为每个值加 2,也就是说,中轴线向X轴正方向移动 2 个单位,得到一个新的数据集。
# 第三个样本中表达量每个值加2
sample3=rnorm(gene_num*cell_num)+2;dim(sample3)=c(gene_num,cell_num)
sample_all=cbind(sample_all,sample3)
colnames(a3)=c(paste0('cell_01_',1:nc),paste0('cell_02_',1:nc))
rownames(a3)=paste('gene_',1:ng,sep = '')
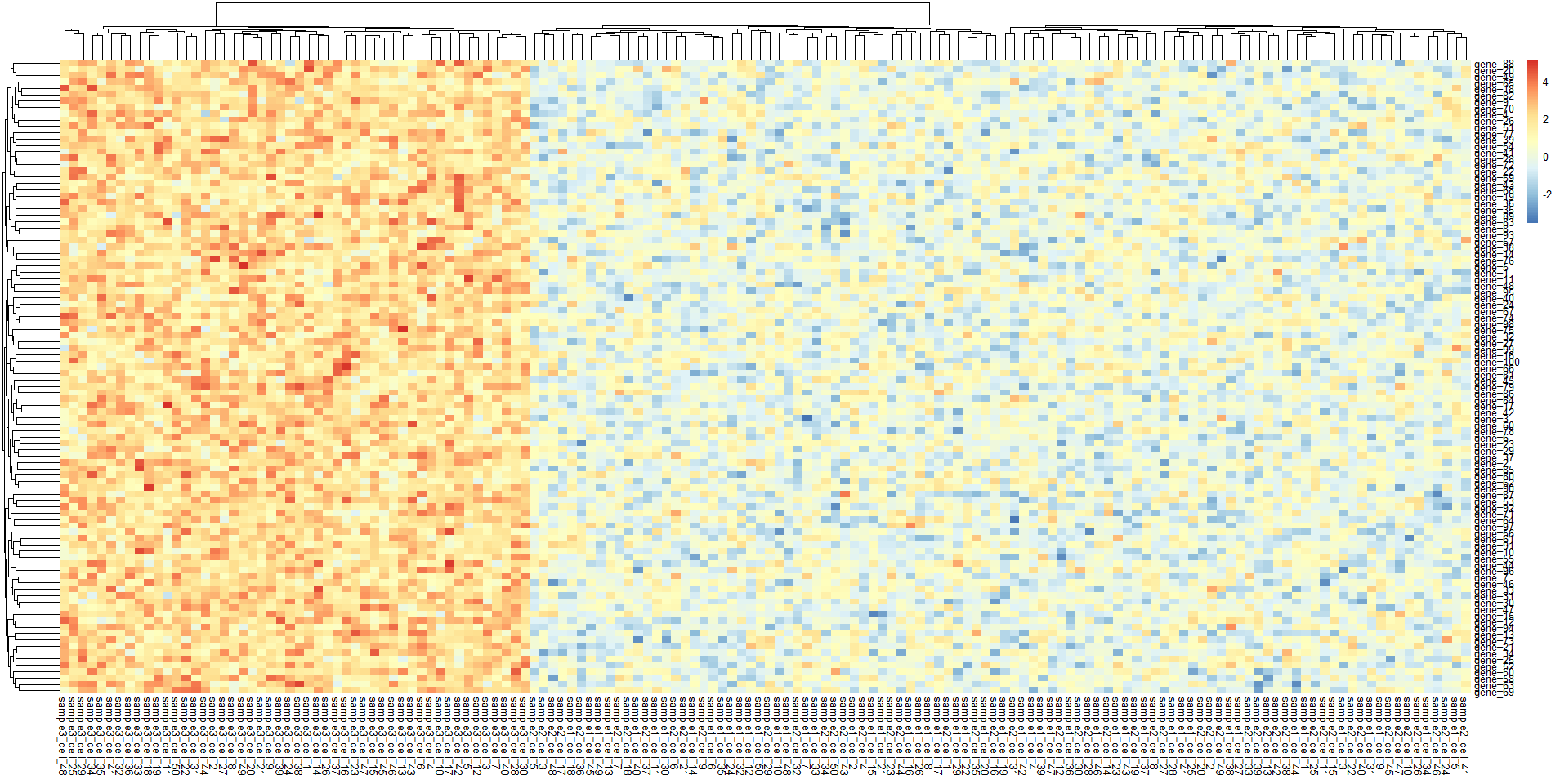
pheatmap(sample_all)
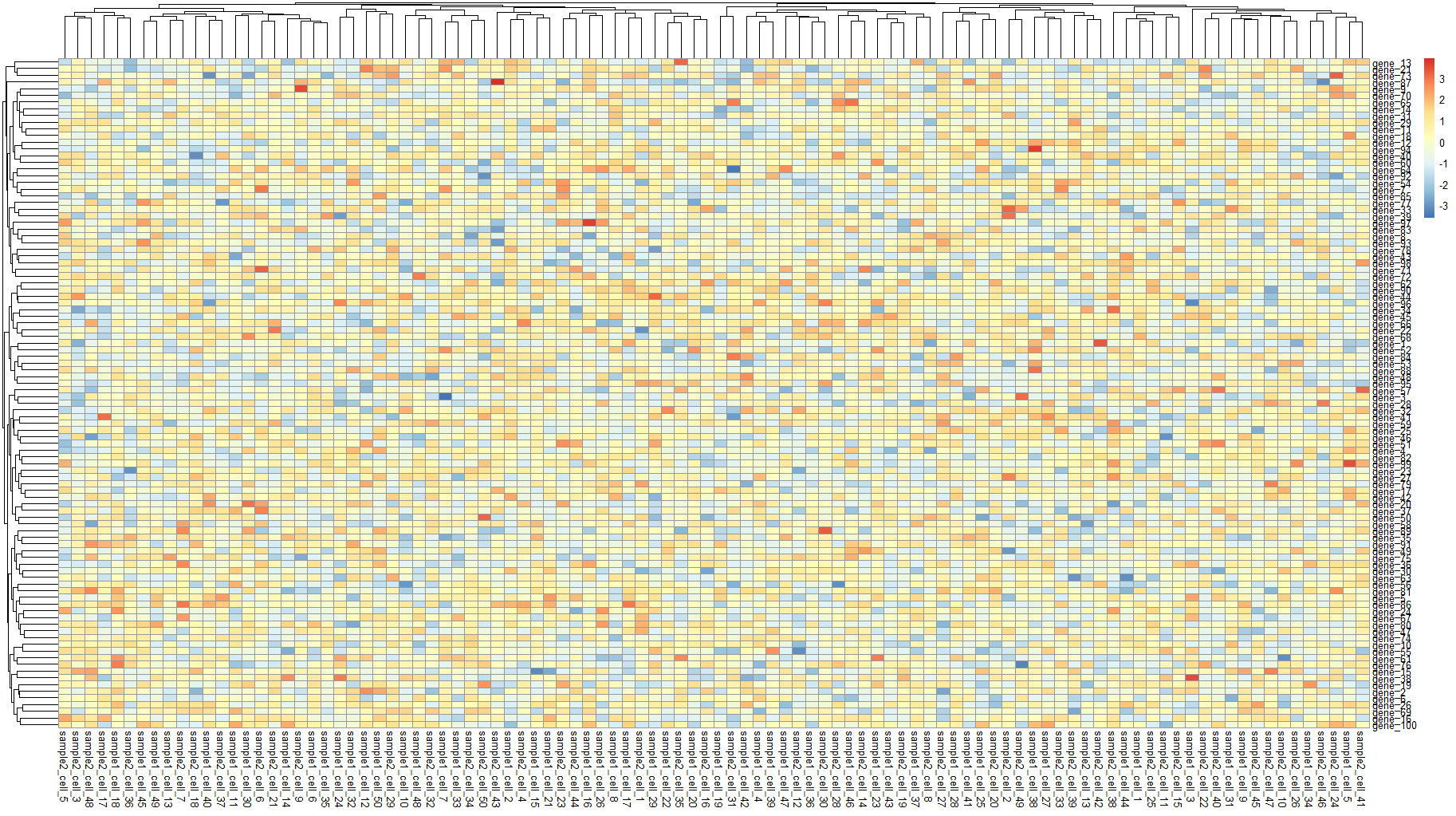
绘制热图,可以明显发现第三个数据集区别与我们之间构建的那两个数据集。这种图应用于实际中就显示出该组数据的异质性。比如,左图是癌症患者的某些基因表达值,区别于右边两个患者的正常表达基因。
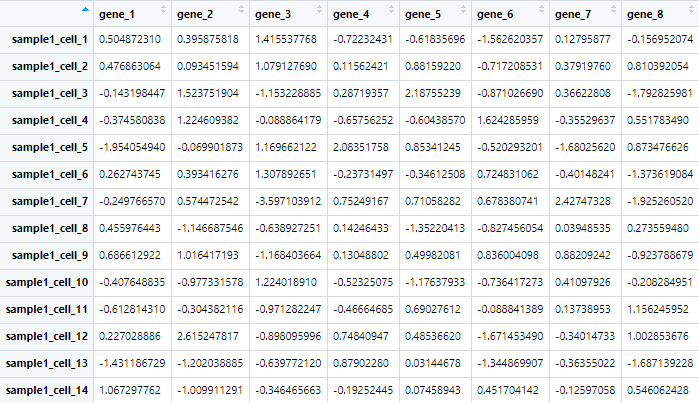
sample_all=t(sample_all);
dim(sample_all)
df=cbind(as.data.frame(a3),group=c(rep('group1',20),rep('group2',20)))
autoplot(prcomp( df[,1:(ncol(df)-1)] ), data=df,colour = 'group')+theme_bw()
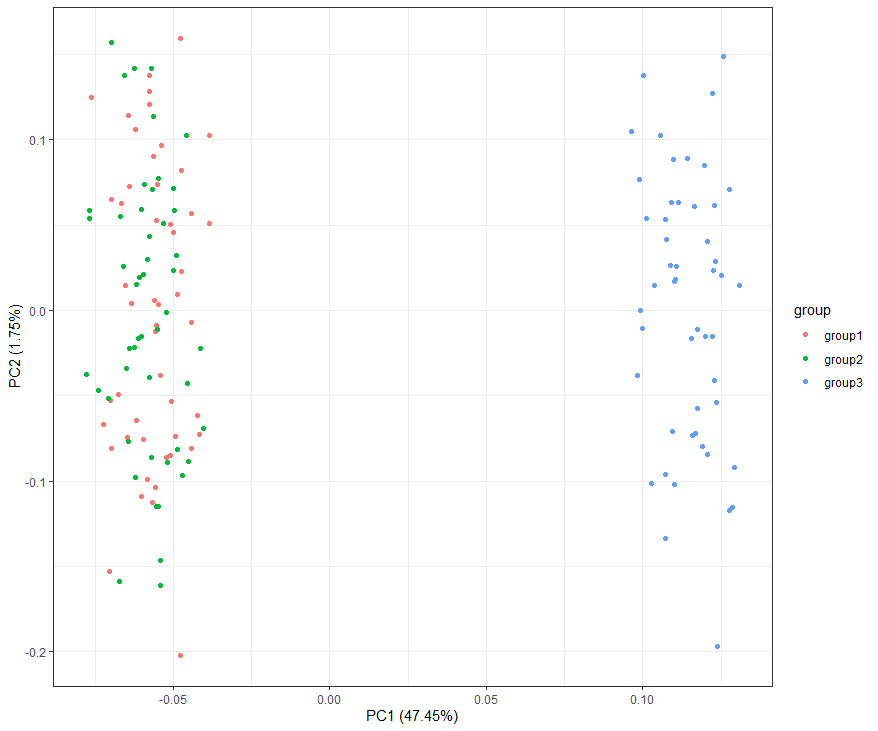
PCA 后我们可以看到,分组极其明显。与前两组相比,新构建的第三个数据集明显分在另一组。比如,该图用于查看测序数据组间是否有差异,样本是否有污染等。而且由于是根据距离公式计算,因此在图中,点之间的距离一定程度也展示了组内或组间不同样本的相似性或异质性。
set.seed(123123)
tsne_out <- Rtsne(sample_all,pca=FALSE,perplexity=10,theta=0.0)
tsnes=tsne_out$Y
colnames(tsnes) <- c("tSNE1", "tSNE2")
tsnes=as.data.frame(tsnes)
group=c(rep('group1',cell_num),rep('group2',cell_num),rep('group3',cell_num))
tsnes$group=group
ggplot(tsnes, aes(x = tSNE1, y = tSNE2))+ geom_point(aes(col=group))
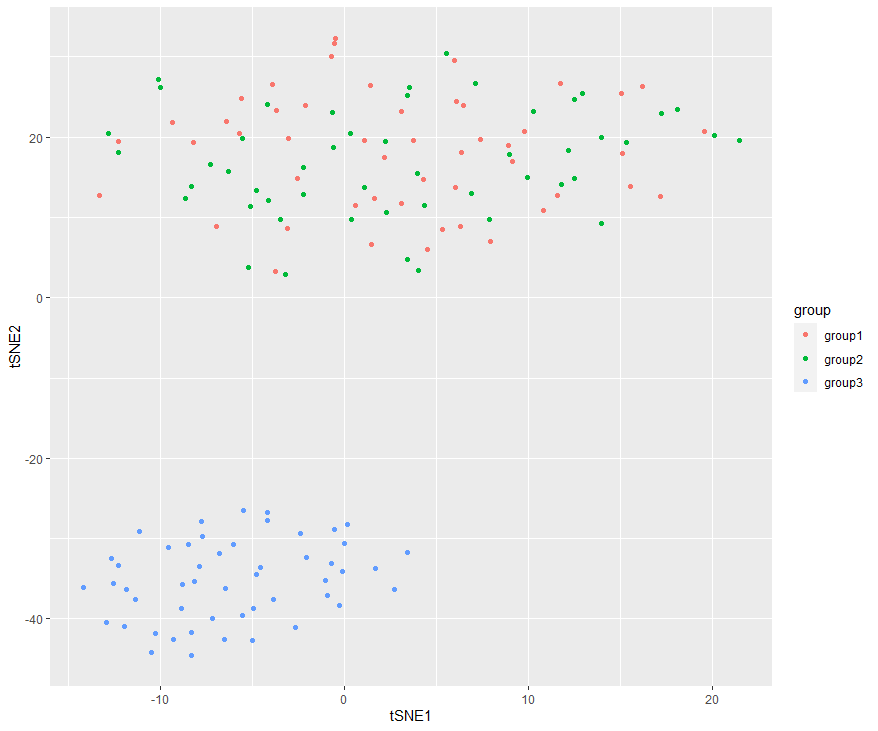
TSNE 图与 PCA 图同样展示出三组样本间的差异。一般用于单细胞测序的细胞聚类,但是与 PCA 不同的是,TSNE 图一般是由三维空间映射到二维平面的成像,所以,组间的距离并不能真实反映样本数据间的差异性。
rm(list=ls())
library(pheatmap)
library(Rtsne)
library(mvtnorm)
# 生成数据(随机)================================
# 设置基因数和细胞数
gene_num=100
cell_num=50
# 设置两个正态分布的随机矩阵(100*50) 作为两个样本矩阵
sample1=rnorm(gene_num*cell_num);
# 生成 100 基因,50 细胞的表达矩阵
dim(sample1)=c(gene_num,cell_num)
# 生成第二个样本
sample2=rnorm(gene_num*cell_num);dim(sample2)=c(gene_num,cell_num)
# 将两个样本合并为一个表达矩阵
sample_all=cbind(sample1,sample2)
# 添加列名
colnames(sample_all)=c(paste0('sample1_cell_',1:cell_num),
paste0('sample2_cell_',1:cell_num))
# 添加行名
rownames(sample_all)=paste('gene_',1:gene_num,sep = '')
# 绘制热图==============================================
pheatmap(sample_all)
# 绘制pca===============================================
# 转置,让行为样本
sample_all=t(sample_all)
# 添加样本分组信息
df=cbind(as.data.frame(sample_all),group=c(rep('group1',cell_num),rep('group2',cell_num)))
# prcomp()主成分分析
pca_dat <- prcomp( df[,1:(ncol(df)-1)] )
autoplot(pca_dat, data=df,colour = 'group')+theme_bw()
# 绘制tsne=============================================
# 进行tSNE
set.seed(123123)
sample_all=t(sample_all)
tsne_out <- Rtsne(sample_all,pca=FALSE,perplexity=10,theta=0.0)
# 获取tSNE的坐标值
str(tsne_out)
# 其中在Y中存储了绘制图坐标
tsnes=tsne_out$Y
colnames(tsnes) <- c("tSNE1", "tSNE2") #为坐标添加列名
# 在此基础上添加颜色分组信息,首先还是将tsnes这个矩阵变成数据框,然后增加一列group信息,最后映射在geom_point中
tsnes=as.data.frame(tsnes)
group=c(rep('group1',cell_num),rep('group2',cell_num))
tsnes$group=group
ggplot(tsnes, aes(x = tSNE1, y = tSNE2))+ geom_point(aes(col=group))
dev.off()
dev.new()
# 构建随机数据集==================================================
# 第三个样本中表达量每个值加2
sample3=rnorm(gene_num*cell_num)+2;dim(sample3)=c(gene_num,cell_num)
colnames(sample3)=c(paste0('sample3_cell_',1:cell_num))
rownames(sample3)=paste('gene_',1:gene_num,sep = '')
sample_all=cbind(sample_all,sample3)
# 绘制热图 =========================================================
pheatmap(sample_all)
# 绘制PCA ==========================================================
sample_all=t(sample_all);
dim(sample_all)
df=cbind(as.data.frame(sample_all),group=c(rep('group1',cell_num),rep('group2',cell_num),rep('group3',cell_num)))
autoplot(prcomp( df[,1:(ncol(df)-1)] ), data=df,colour = 'group')+theme_bw()
# 绘制tSNE=========================================================
set.seed(123123)
tsne_out <- Rtsne(sample_all,pca=FALSE,perplexity=10,theta=0.0)
tsnes=tsne_out$Y
colnames(tsnes) <- c("tSNE1", "tSNE2")
tsnes=as.data.frame(tsnes)
group=c(rep('group1',cell_num),rep('group2',cell_num),rep('group3',cell_num))
tsnes$group=group
ggplot(tsnes, aes(x = tSNE1, y = tSNE2))+ geom_point(aes(col=group))
本文为二次转载,如有侵权请联系删除。
文章
12.03W+人气
19粉丝
1关注
更多数字孪生可视化干货内容






 扫一扫关注公众号
扫一扫关注公众号
 扫一扫联系客服
扫一扫联系客服
©Copyrights 2016-2022 杭州易知微科技有限公司 浙ICP备2021017017号-3  浙公网安备33011002011932号
浙公网安备33011002011932号
互联网信息服务业务 合字B2-20220090







